La legge di Raoult fu elaborata nel 1886 dal chimico Franςois-Marie Raoult ed è utilizzata per descrivere gli equilibri liquido-vapore. Nello specifico tale legge descrive la variazione della tensione di vapore di un solvente una volta che viene aggiunto un soluto.
Secondo la Legge di Raoult, la tensione di vapore di una soluzione è direttamente proporzionale alla frazione molare del solvente. Tale legge può essere espressa sottoforma di equazione:
Psoluzione = xsolvente · P°solvente
Psoluzione = tensione di vapore della soluzione
P°solvente = tensione di vapore del solvente puro a quella specifica temperatura
xsovente = frazione molare del solvente
Se la frazione molare del solvente (xsovente) è uguale a 1, allora la soluzione è costituita solo dal solvente e la tensione di vapore della soluzione coincide con quella del solvente.
Se nella soluzione viene aggiunta una certa quantità di soluto non volatile allora si può ricavare la variazione della tensione di vapore:
ΔP = xsoluto · P°solvente
Tanto maggiore è la frazione molare di soluto non volatile in soluzione, tanto più marcata è la diminuzione di tensione di vapore della soluzione.
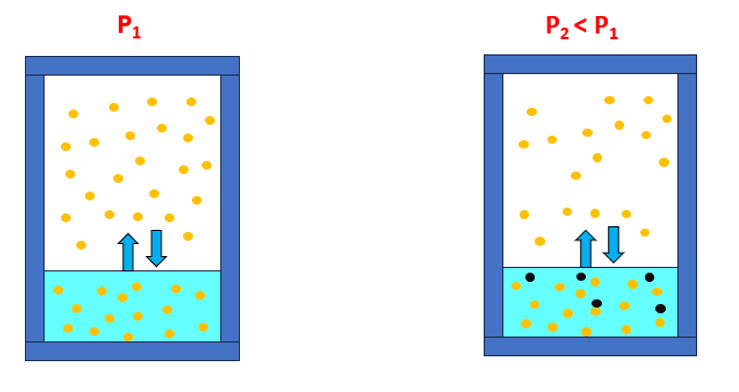
Da un punto di vista fisico questo fenomeno può essere spiegato considerando la distribuzione delle particelle di soluto una volta sciolto nella soluzione. Queste si distribuiscono in maniera omogenea nel volume di solvente e una parte di esse si distribuisce anche sulla superficie libera. L’evaporazione è un fenomeno che non coinvolge l’intera massa di liquido ma solo la parte superficiale. Queste particelle di soluto rendono più difficoltosa l’evaporazione delle molecole di solvente, comportando un abbassamento della tensione di vapore della soluzione (Figura 1). Il fenomeno inverso, ovvero la condensazione, non è invece in alcun modo intaccato dal soluto.
Si raggiunge un nuovo equilibrio con valori di tensione di vapore saturo inferiori rispetto a quelli del liquido puro. Tanto maggiore è la quantità di soluto tanto più tale effetto è marcato.
Figura 1 – Immagine di due recipienti: a sinistra è presente solo il solvente puro, a destra anche il soluto non volatile che rende più difficoltosa l’evaporazione al solvente.
Cosa succede quando la mia soluzione è costituita da due o più componenti volatili?
In questo caso ciascun componente contribuisce alla pressione di vapore totale con la propria pressione parziale. In una soluzione costituita da due componenti volatili, il vapore che si viene a formare è formato in parte dal primo componente e in parte dal secondo. Il contributo di ciascun componente è tanto maggiore quanto più esso è presente nella soluzione.
Ptot = Pa + Pb
La tensione di vapore totale della soluzione è data dalla somma delle pressioni parziali dei singoli componenti.
Pa = xaPa°
Pb = xbPb°
Ptot = xaPa° + xbPb°
Poiché:
xa + xb =1
Ptot = xaPa° + (1 – xa) Pb°
Da cui si ricava:
Ptot = Pb° + (Pa°-Pb°) xa
Le soluzioni che rispondono a questa equazione si definiscono soluzioni ideali, ovvero soluzioni in cui le molecole dissimili (A-B) hanno lo stesso livello d’interazioni tra le molecole uguali (A-A e B-B). Un ottimo esempio di soluzione ideale è il sistema benzene-toluene
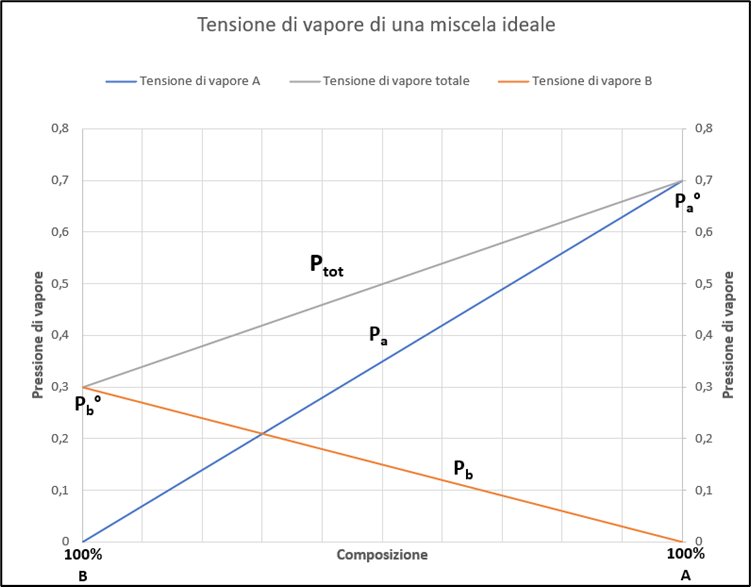
Per questo tipo di sistemi è possibile costruire un diagramma (Figura 2) in cui la pressione totale è data dalla somma delle pressioni dei singoli componenti. Da questo diagramma è possibile ricavare la pressione parziale dei singoli componenti e la pressione totale al variare della composizione.
Figura 2 – Andamento delle pressioni parziali e della pressione totale al variare della composizione per una miscela ideale.
DEVIAZIONE DAL COMPORTAMENTO IDEALE
Non tutte le soluzioni mostrano un comportamento ideale. Infatti, in alcuni sistemi il livello d’interazione tra le molecole dissimili (A-B) è minore rispetto alle molecole uguali (A-A e B-B). Un esempio è la soluzione costituita da acetone e disolfuro di carbonio. Qualora l’intorno di una molecola A sia costituito da molecole B, la minore interazione A-B facilita il processo di evaporazione. Considerando la legge di Raoult:
Pa > xaPa° e Pb > xbPb°
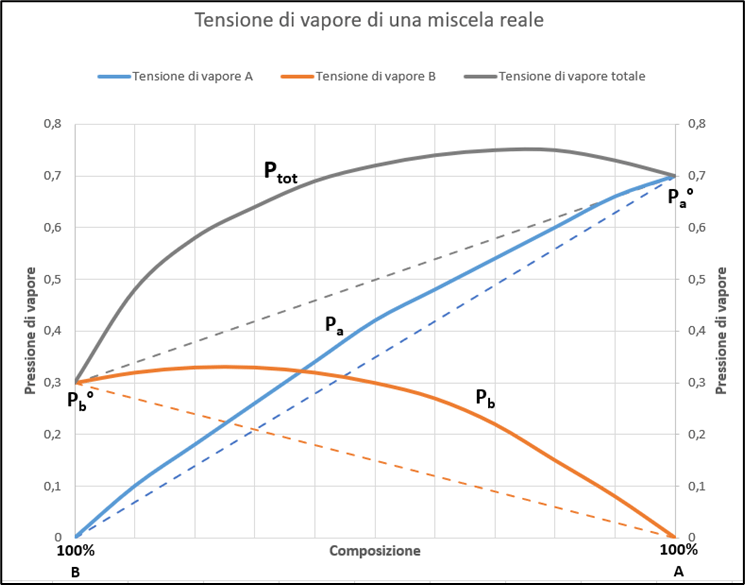
Queste deviazioni positive della tensione di vapore rispetto a quelle calcolate dalla legge di Raoult possono essere rappresentate nel diagramma in Figura 3. Da sottolineare che i singoli valori sono determinati sperimentalmente e possono variale a seconda dei componenti della soluzione.
Figura 3 – Deviazioni positive delle pressioni parziali e della pressione totale al variare della composizione per una miscela reale.
Alcuni sistemi esibiscono un livello d’interazione tra le molecole dissimili (A-B) maggiore rispetto alle molecole uguali (A-A e B-B). Un esempio è la soluzione costituita da acqua ed etanolo. Qualora l’intorno di una molecola A sia costituito da molecole B, la maggiore interazione A-B renderà più difficoltoso il processo di evaporazione. Considerando la legge di Raoult:
Pa < xaPa° e Pb < xbPb°
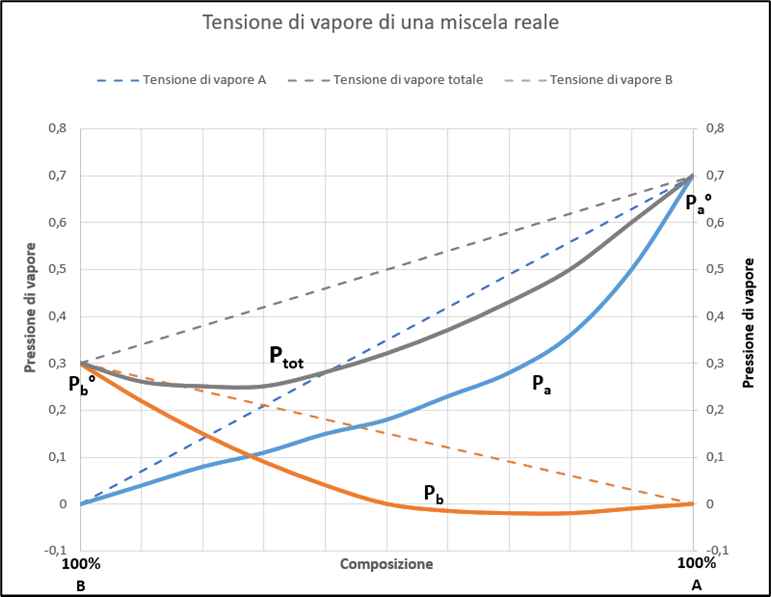
Queste deviazioni negative della tensione di vapore rispetto a quelle calcolate dalla legge di Raoult possono essere rappresentate nel diagramma in Figura 4. Analogamente a quanto riportato sopra, i singoli valori sono determinati sperimentalmente e possono variale a seconda dei componenti della soluzione.
Figura 4 – Deviazioni negative delle pressioni parziali e della pressione totale al variare della composizione per una miscela reale.
CONCETTI CHIAVE:
- La legge di Raoult governa gli equilibri liquido-vapore.
- La tensione di vapore di una soluzione diminuisce quando viene aggiunto un soluto non volatile.
- Tale diminuzione è proporzionale alla frazione molare di tale soluto.
- In presenza di più componenti in soluzione, la tensione di vapore della soluzione è dato dalla somma delle tensioni di vapore dei singoli componenti.
- Esistono delle deviazioni negative e positive della tensione di vapore rispetto a quelle determinate dalla legge di Raoult.


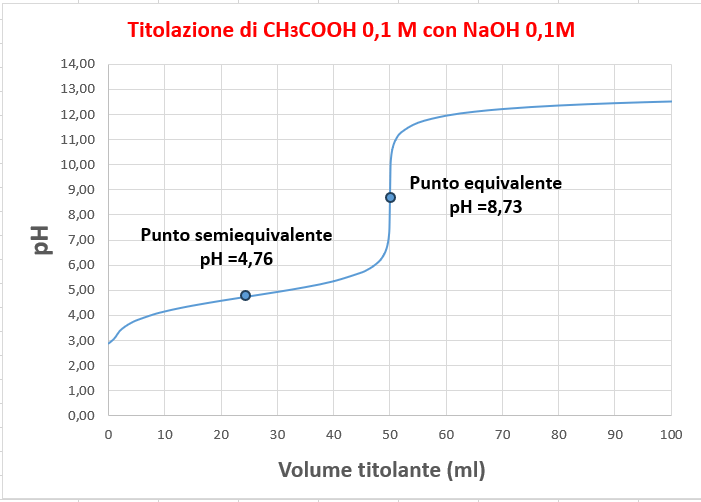
![pH = pK_{a} + log_{10}\frac{[CH_{3}COO^{-}]}{[CH_{3}COOH]}](https://s0.wp.com/latex.php?latex=pH+%3D+pK_%7Ba%7D+%2B+log_%7B10%7D%5Cfrac%7B%5BCH_%7B3%7DCOO%5E%7B-%7D%5D%7D%7B%5BCH_%7B3%7DCOOH%5D%7D&bg=ffffff&fg=000000&s=2&c=20201002)

![pH = 14-(-log_{10}(\sqrt{K_{b}\cdot [CH_{3}COO^{-}]})](https://s0.wp.com/latex.php?latex=pH+%3D+14-%28-log_%7B10%7D%28%5Csqrt%7BK_%7Bb%7D%5Ccdot+%5BCH_%7B3%7DCOO%5E%7B-%7D%5D%7D%29&bg=ffffff&fg=000000&s=2&c=20201002)
![[OH^{-}] =\frac{C_{NaOH}V_{NaOH}\;-\;C_{CH_{3}COOH}V_{CH_{3}COOH}}{V_{HCl}\;+\;V_{NH_{3}}}](https://s0.wp.com/latex.php?latex=%5BOH%5E%7B-%7D%5D+%3D%5Cfrac%7BC_%7BNaOH%7DV_%7BNaOH%7D%5C%3B-%5C%3BC_%7BCH_%7B3%7DCOOH%7DV_%7BCH_%7B3%7DCOOH%7D%7D%7BV_%7BHCl%7D%5C%3B%2B%5C%3BV_%7BNH_%7B3%7D%7D%7D&bg=ffffff&fg=000000&s=2&c=20201002)